






 Official publication of the Czech Society of Ultrasound in Obstetrics and Gynecology.
Official publication of the Czech Society of Ultrasound in Obstetrics and Gynecology.



Uterine endometrioid cancer represents a significant gynecological malignancy with a rising global incidence. Using RNA sequencing,in our pilot study we identified 2,483 differentially expressed genes, comprising protein-coding genes, genes for non-coding RNAs, and pseudogenes, in tumor tissues compared to healthy counterparts. In our study we focused on comparism of proteing-coding genes. Principal Component Analysis revealed clustering based on histological grade. Pathway analysis highlighted the downregulation of Wnt and AGE-RAGE signaling, alongside the upregulation of cell cycle regulation pathways. These findings provide molecular insights into endometrioid cancer and suggest potential biomarkers and therapeutic targets for improved management strategies.
Endometrioid cancer is one of the most prevalent gynecological malignancies, with rising incidence attributed to factors such as obesity, hormonal imbalances, and excessive estrogen exposures typically develops in the uterine lining and is associated with endometrial hyperplasia as a precursor lession (1). The non-specific symptoms of early-stage endometrial cancer, such as abnormal uterine bleeding, often delay diagnosis (2). Consequently, there is an urgent need for reliable biomarkers and targeted therapies to improve diagnostic accuracy and patient outcomes (3). Molecular profiling, including transcriptomics, has revolutionized our understanding of cancer biology. Transcriptomic profiling offers insights into local gene expression and is viable approach to the study of the functional impact of genetic variations. It can identify differentially expressed genes (DEGs) and understand their role in tumor development, progression, and therapeutic resistance (4,5).
Uterine endometrioid cancer particularly is suited for transcriptomic analysis due to its heterogeneous nature and the influence of hormonal pathways on tumor biology. Dysregulated signaling pathways, the Wnt, PI3K/AKT, and p53 pathways, have been identified as critical players in its pathogenesis. In addition, studies have shown that immuneresponse related features and the tumor microenvironment significantly affect disease progression, further underscoring the need for molecular characterization (6).
Despite advances in research, many challenges remain, presenting a significant gap in the identification of stagespecific biomarkers and pathways that could inform early detection and potential personalized treatment. Furthermore, the integration of transcriptomic data with proteomics and metabolomics could provide a more comprehensive understanding of the disease (7). This study leverages transcriptomic analysis to uncover dysregulated pathways, providing insights for understanding behavior of development, procession, mangemetn and potencialy novely therapeutic strategies in endometrial carcinoma.
We analyzed 12 patients diagnosed with uterine endometrioid cancer. Patients were enrolled in the BIOMEDIRES 2 study in Slovakia, written informed consent was obtained from all participants, the study was approved by the ethics committee. The project was based on a multidisciplinary approach in the analysis of potential endometrial tumor markers. The number of participants in the study is limited by data collection from only one workplace and is ultimately influenced by the selection inclusion criteria for transcriptomic analysis.
The age range of the patients in selected cohort was 49-74 years, with a mean age of 61.4 years. All tumours were confirmed by endometrioid histology, which 7 patients were FIGO stage 1a and 5 were FIGO stage 1b. By FIGO grading 8 patients were G1, 5 patients G2 and one patient G3 respectivelly.
The analyzed tissue came from tumor samples taken before a rapid preoperative biopsy performed during the operation itself, with subsequent comparison to the patient‘s healthy endometrial tissue. Approximately 25 mg samples of both uterine cancer and healthy tissues were immersed in DNA/RNA Shield.
Tissue samples underwent RNA extraction and sequencing using Illumina‘s NextSeq 500 platform. Statistical analysis to identify the differentially expressed genes was done on per gene read counts outputs of RNA-STAR, while both normalization and statistical test was performed by Deseq2 v.1.38.3. Genes (with p ≤ 0.05, log2 Fold Change > 1) were annotated using R package biomaRt v.2.58.2.
Pathway analysis was performed using R instance of gProfiler2 v.0.2.1. “Over-expressed” and “under-expressed” gene lists (under condition of adjusted p-value lower than 0.05, and |log2 Fold Change| > 1) were used as a query for the analysis (ranked according to p-values). The set of all genes expressed in at least one of our samples were set as custom background. KEGG, Reactome, and Gene Ontology database pathways were used.
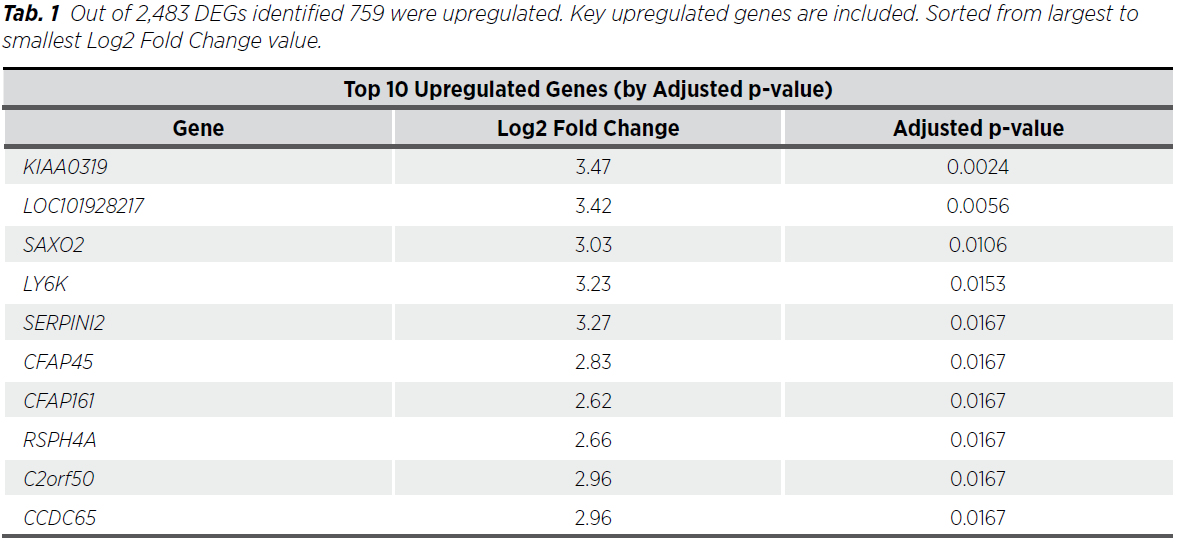
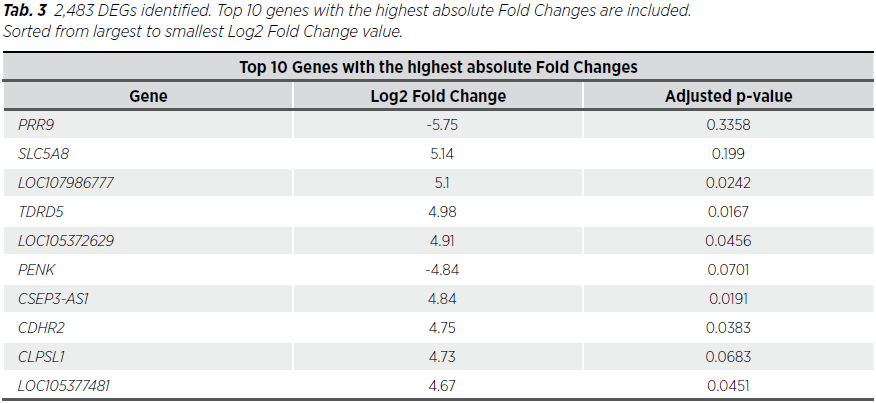
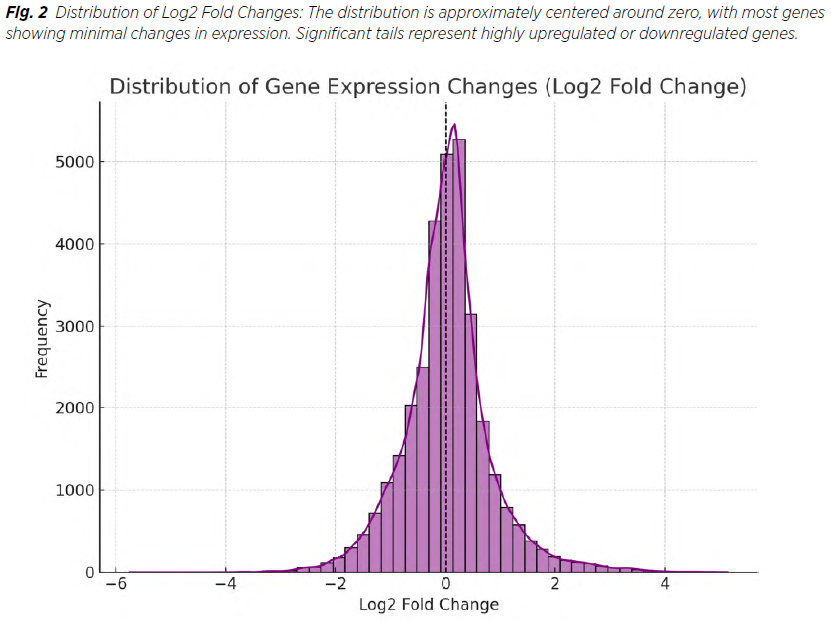
Out of 2,483 dysregulated genes (DEGs) identified, 1,724 were downregulated and 759 upregulated. Key upregulated genes included KIAA0319 and LOC101928217, while downregulated genes included APOLD1. Overall, genes with the highest absolute Fold Changes include PRR9 and SLC5A8 (Tab. 1, 2, 3) and globally Figure 1, 2 .
The results highlight key targets for further investigation and follow-up in endometrioid carcinoma and draw attention to the potential use of the biomarkers KIAA0319 and APOLD1, which were found to have significantly different gene expression.
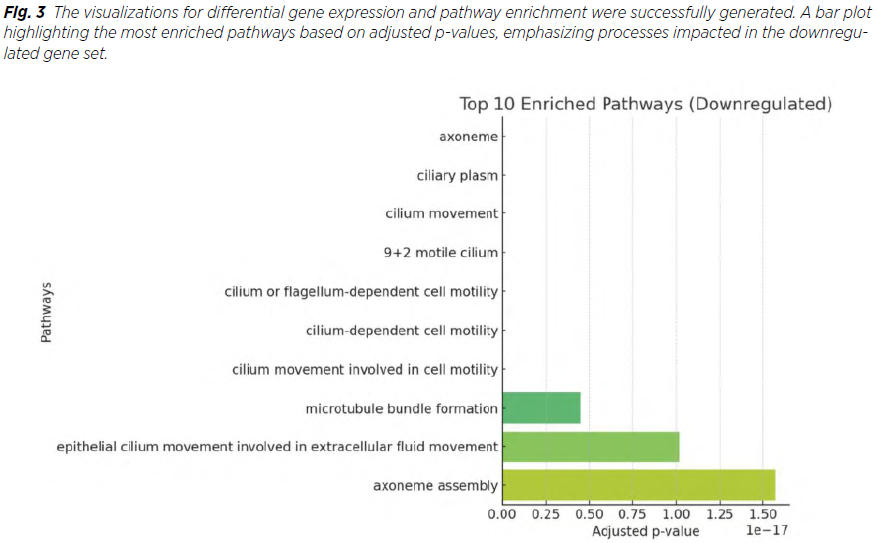
KEGG analysis highlighted:
Our study identified several enriched KEGG pathways, which are also in line with previous research findings. Visualization includes pathway enrichment maps (Fig. 3) .
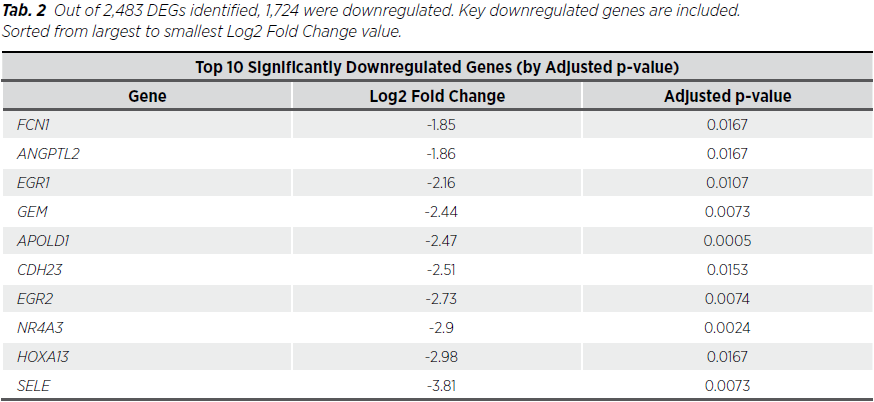
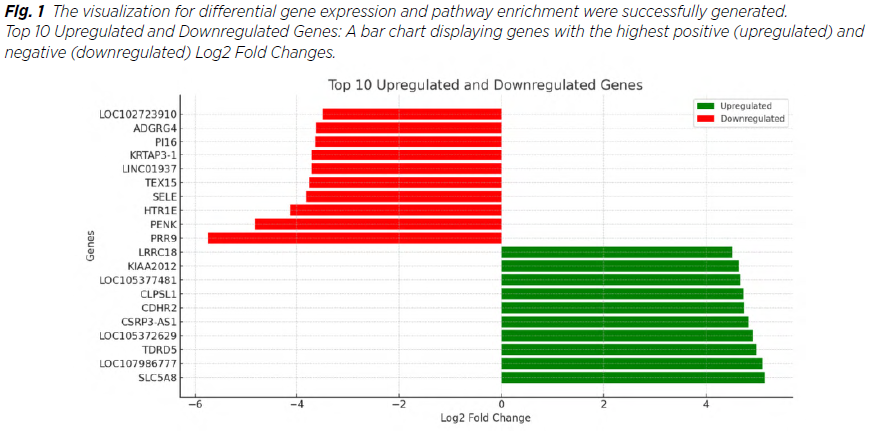
The findings from this study align with and expand upon existing research in uterine endometrioid cancer, particularly regarding the dysregulation of critical cellular pathways (8). The identification of 2,483 differentially expressed genes (DEGs) underscores the molecular complexity of this malignancy, with specific genes showing significant potential as diagnostic biomarkers or therapeutic targets (9). For instance, the upregulation of KIAA0319 and LOC101928217 may indicate their roles in tumor progression, while the downregulation of APOLD1 could reflect disrupted vascular integrity or reduced angiogenesis in tumor microenvironments. Low expression of HOXA13 is associated with poorer survival and has potential as a prognostic biomarker. Downregulation of several other of these genes has also been observed in various malignancies: APOLD1 in seminoma and embryonal carcinoma, NR4A3 in all subtypes of breast carcinoma. These molecular alterations offer a foundation for further exploration of their functional roles in endometrial cancer biology (10).
The dysregulation of the Wnt signaling pathway, observed in this study, corroborates its established role in epithelial tumorigenesis. Wnt signaling is integral to cell proliferation, migration, and differentiation, and its downregulation suggests altered cellular communication within tumor tissue (11). On the other hand, the upregulation of cell cycle pathways emphasizes the aggressive proliferative nature of the cancer, aligning with findings in other malignancies where cell cycle dysregulation is a hallmark of tumor progression. The identification of the AGE-RAGE signaling pathway as downregulated highlights its dual role in cancer. This pathway is known for mediating inflammatory responses and promoting oxidative stress, which are critical in cancer initiation. Its downregulation in this context could indicate a shift in the tumor‘s reliance on alternative prosurvival and inflammatory pathways . Downregulation of pathways associated with vascular smooth muscle contraction, cGMP-PKG signaling, AGE-RAGE signaling in diabetic complications, dilated cardiomyopathy, protein digestion and absorption, ECM-receptor interaction, resistance to EGFR tyrosine kinase inhibitors, growth hormone synthesis, secretion and action, Wnt signaling pathway, phospholipase D signaling pathway, regulation of lipolysis in adipocytes, apelin signaling pathway and relaxin signaling pathway was found. Our results are also in line with upregulation of cell cycle, biosynthesis of mucin-type O-glycans, amino acid biosynthesis and glutathione metabolism. These insights provide a broader understanding of how uterine endometrioid cancer adapts molecularly to its microenvironment (10,12).
Furthermore, the high absolute Fold Changes observed in SLC5A8 and PRR9 suggest that these genes may also serve as therapeutic targets or indicators of treatment response, meriting further functional studies (13,14). The SLC5A8 gene is downregulated in various types of malignancies, including cervical cancer. The results of this year‘s study in this area demonstrated that the SLC5A8 gene is downregulated in cervical cancer by hypermethylation of a CpG island in the gene promoter (15,16).
The integration of transcriptomic data with clinical information could pave the way for precision medicine approaches in endometrial cancer. For example, combining RNA-seq results with immunohistochemical analyses or liquid biopsy techniques could enhance the detection of disease-specific biomarkers and monitor disease progression in real time. Additionally, multi-omics approaches, including proteomics and epigenomics, could reveal deeper insights into gene regulation and pathway interactions that are not evident from transcriptomic data alone (17).
Finally, we have to highlight our pilot study limitation and its impact on the generalizability of our findings, while emphasizing the importance of these preliminary results in generating hypotheses for larger-scale studies.
Finally, this study highlights the importance of further research into the molecular mechanisms underlying tumor progression in endometrioid cancer. Future investigations should focus on longitudinal studies to validate these findings in larger cohorts and explore their relevance in metastatic or recurrent settings. The identification of stage-specific biomarkers and treatment targets could significantly improve the management and survival outcomes of patients with this disease. Overall, this transcriptomic analysis contributes to the growing body of knowledge on uterine endometrioid cancer, emphasizing its molecular complexity and identifying promising avenues for further research and clinical application.
