





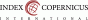
 Rada pro výzkum, vývoj a inovace: Seznam recenzovaných neimpaktovaných časopisů vydávaných v ČR
Oficielní časopis České společnosti pro ultrazvuk v porodnictví a gynekologii.
Rada pro výzkum, vývoj a inovace: Seznam recenzovaných neimpaktovaných časopisů vydávaných v ČR
Oficielní časopis České společnosti pro ultrazvuk v porodnictví a gynekologii.



Problematika trombotických mikroangiopatií (TMA) představuje, nejen v porodnictví, velmi závažný patologický stav, který je spojen s tvorbou trombóz na úrovni kapilár i arteriol v důsledku poškození endotelu a aktivace komplementu. Je provázen mikroangiopatickou hemolytickou anémií (MAHA), trombocytopenií a dysfunkcí různých orgánů. Relativně často je navíc spojen se sekundárními systémovými změnami srážlivosti. TMA zahrnují velmi nesourodou skupinu syndromů a stavů, kdy ke konečné diagnóze docházíme postupným vylučováním jednotlivých příčin („per exclusionem“). V porodnické praxi se nejčastěji setkáváme s tím, že těhotné/rodičky/nedělky se prezentují pod obrazem preeklampsie/HELLP syndromu (hemolysis, elevated liver enzymes, low platelets). Tento všem porodníkům jinak dobře známý stav zahrnuje obraz MAHA (dynamické snižování hladiny hemoglobinu, zvyšování hladiny bilirubinu, snížení haptoglobinu, přítomnost schistocytů v periferním nátěru krve), periportální ischémie jater (elevace transamináz) a trombocytopenie v důsledku vyšší agregace trombocytů v poškozené periferní mikrocirkulaci. HELLP syndrom se řadí mezi TMA také, měl by však spontánně odeznívat přibližně do 48-72 hodin po porodu. Pakliže se tak nestane, je velmi důležité pomýšlet na jiné příčiny TMA, které často představují ještě vážnější ohrožení života než HELLP syndrom. Důkladná znalost diferenciální diagnostiky je proto velmi důležitá. Problematiku tedy musí dobře ovládat každý poskytovatel zdravotní péče těhotným ženám, a proto ji kolektiv autorů předkládá ve formě tohoto doporučeného postupu.
Problematika trombotických mikroangiopatií (TMA) představuje, nejen v porodnictví, velmi závažný patologický stav, který je spojen s tvorbou trombóz na úrovni drobných cév (kapilár a arteriol) a se současným poškozením endotelu a aktivací komplementu. TMA je charakterizována klasickou triádou (1,2):
• mikroangiopatickou hemolytickou anémií (MAHA); charakteristické pro ni je rychlé snižování hladiny hemoglobinu (Hb) pod 100 g/l a vysoká aktivita laktátdehydrogenázy (LD) nad 1,5násobek horní hranice normy, přítomnost schistocytů v periferním nátěru krve (fragmenty erytrocytů, které vznikají mechanickým poškozením v krevním oběhu), snížení koncentrace haptoglobinu (následně hemopexinu), zvýšení koncentrace bilirubinu a negativní přímý antiglobulinový test (odpovídá neimunitní, mechanické hemolýze);
• trombocytopenií (< 150 x 109/l nebo poklesem počtu trombocytů o > 25 % proti hodnotě před rozvojem trombotické mikroangiopatie);
• dysfunkcí různých orgánů v důsledku jejich ischemizace.
Důležité je mít na paměti, že v řadě případů TMA se můžeme setkat s nekompletní manifestací výše uvedených příznaků, včetně chybění trombocytopenie. Relativně často jsou tyto stavy spojeny se sekundárními systémovými změnami srážlivosti krve.
Projevy TMA doprovázejí řadu velmi nesourodých stavů či chorob, kdy ke konečné diagnóze docházíme postupným vylučováním jednotlivých příčin („per exclusionem“).
Nejdůležitější a nejčastější jednotky spojené s TMA jsou:
• trombotická trombocytopenická purpura (TTP), hereditární či získaná;
• TMA asociované s těhotenstvím – preeklampsie (PE), HELLP syndrom (hemolýza, elevované jaterní enzymy a trombocytopenie), AFLP (acute fatty liver of pregnancy, akutní těhotenská steatóza);
• s infekcí asociovaný hemolyticko-uremický syndrom (HUS), kam patří zejména STEC-HUS (způsobený infekcí Escherichia coli produkující shiga toxin), neuraminidázový HUS (při pneumoniích způsobených kmeny Streptococcus pneumonie produkujícími neuraminidázu) či při infekcích jako HIV, influenza, ale i SARS-CoV-2;
• atypický HUS (aHUS), který se dnes doporučuje označovat jako komplementem mediovaný HUS (CM-HUS) – základní roli zde hraje dysregulace alternativní cesty komplementu;
• HUS způsobený deficitem kobalaminu a HUS při mutacích v genu pro DGKE (diacylglycerol kináza ε);
• sekundární formy TMA/HUS (sHUS) doprovázející řadu patologických stavů (nádory, maligní hypertenze, autoimunitní choroby, stavy po transplantaci solidních orgánů či kostní dřeně) či spojené s podáváním léků (některá cytostatika, imunosupresiva, antiagregancia).
Těhotenství a peripartální období představuje vysoce rizikové situace, které často vedou k rozvoji TMA. Během nich se můžeme setkat zejména se čtyřmi nejdůležitějšími formami TMA, a sice: TTP, PE/HELLP syndromem, CM-HUS a antifosfolipidovým syndromem (APS) (3). Těhotné/rodičky/ nedělky se nejčastěji prezentují pod obrazem PE/HELLP syndromu. Tento všem porodníkům jinak dobře známý stav by měl spontánně odeznívat do 48-72 hodin po porodu. Pakliže se tak nestane, je velmi důležité pomýšlet na jiné příčiny TMA, které často představují ještě vážnější ohrožení života, než je HELLP syndrom. Rozvoj TMA během gravidity 4,5krát zvyšuje riziko mortality oproti těhotným bez TMA (4). Je zde i vyšší riziko morbidity, kdy až 81 % těhotných/ rodiček s TMA vyžaduje dialýzu a skoro polovina progreduje do terminálního renálního selhání (end stage kidney disease - ESKD) (5).
Hlavním účelem tohoto dokumentu je poskytnout zdravotníkům aktuální ucelený pohled na problematiku TMA a seznámit je s diagnostikou a managementem TMA u těhotných a postpartum.
Vzhledem k tomu, že samotný klinický obraz k rozlišení jednotlivých typů TMA nestačí, je třeba pro včasnou diagnózu a vhodnou léčbu využít robustní patofyziologické znaky. V posledních desetiletích se chápání úlohy komplementu v patofyziologii TMA rychle vyvíjelo, což mj. vedlo k lepší charakterizaci onemocnění, která jsou dysregulací komplementu provázena. CM-HUS již dlouho slouží jako model onemocnění, u kterého mutace genů exprimujících různé složky komplementu vedou v konečném důsledku k neregulované aktivaci alternativní cesty komplementu, sekundárnímu poškození endotelu a masivní orgánové ischemizaci s jejich dysfunkcí. Nejvíce je patrné narušení funkce ledvin, jater, srdce a také nespecifické projevy poškození mikrocirkulace mozku – encefalopatie. Lepší pochopení úlohy komplementu u těchto diagnóz však pomohlo najít účinné terapeutické nástroje. Přestože je nezpochybnitelné, že v případě CM-HUS je hlavním patogenetickým podkladem dysregulace alternativní cesty komplementu, pro rozvoj onemocnění je nutná přítomnost tzv. druhého zásahu, spouštěče, který odmaskuje do té doby „spící“ predispozici. Nejčastějšími těmito spouštěči jsou infekce, traumata, těžká hypertenze, těhotenství, a zejména porod. Patří mezi ně i postpartální krvácení (postpartum hemorrhage, PPH). Současně také platí, že čím větší je genetická predispozice, tím i mírnější druhý zásah stačí na to, aby se CM-HUS spustil. Naopak platí to samé; např. velmi silné PPH vede k sekundární aktivaci koagulační kaskády, ale i aktivaci komplementu, a může vést k jeho nekontrolované amplifikaci. Proto PPH s velkou krevní ztrátou mohou vyústit v rozvoj TMA, která se velmi obtížně rozeznává od CM-HUS. Důsledky na úrovni poškozených tkání jsou navíc hodně podobné. To vše může velmi komplikovat identifikaci toho správného onemocnění a diferenciální diagnostiku těchto stavů.
Druhý patofyziologický mechanizmus rozvoje TMA zahrnuje primární poškození endotelu nějakou (endo- či exogenní) noxou a následnou druhotnou aktivaci komplementu. Faktory vedoucí k poškození endotelu zahrnují především bakteriální endotoxiny (Shiga-toxin, neuraminidáza), léky, viry či některá autoimunitní onemocnění (vaskulitidy, systémový lupus erythematodes – SLE). Nespecifická aktivace endotelu s excesivní expresí von Willebrandova faktoru (vWF) může nastartovat akutní ataku vrozené TTP nebo přispět k exacerbaci získané TTP nebo ke klinickému relapsu po dosažení jen parciální remise TTP.
Poslední patofyziologický mechanizmus, který může druhotně vést k rozvoji TMA, zahrnuje stavy primárně postihující koagulační systém (např. katastrofický antifosfolipidový syndrom – CAPS). Vznik intravaskulárních trombů vede k další aktivaci nebo poškození endotelu a současné nadměrné aktivaci komplementu. Výsledkem může být rozvoj MAHA/TMA.
TTP je vzácné, klinicky závažné onemocnění ze skupiny TMA s vysokou mortalitou, pokud není včas zahájena adekvátní léčba. Manifestovat se může v kterékoli fázi těhotenství, nejčastěji to ale bývá ve 3. trimestru. Rozeznáváme získanou formu TTP (imunitně navozenou) a kongenitální (Upshaw-Schülman syndrom). Incidence TTP se odhaduje na 2-3/100 000 gravidit (6). Těhotenství nicméně představuje vysoce rizikový stav pro vznik této choroby; 12-25 % všech TTP v dospělosti se rozvijí právě během těhotenství (7). Pokud se TTP poprvé objeví během těhotenství nebo se jedná o relaps již známé diagnózy, představuje to závažný stav spojený s vysokým rizikem ohrožení matky (multiorgánové selhání) i plodu (riziko fetálního úmrtí).
Příčinou onemocnění je těžký deficit depolymerázy ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13) štěpící multimery vWF. Ve většině případů se jedná o získaný deficit vznikající v důsledku tvorby autoprotilátek proti této metaloproteáze. Vrozený deficit je velmi vzácný a je způsoben mutacemi v genu pro ADAMTS13. Nicméně se ukazuje, že až polovina kongenitálních forem TTP se může manifestovat během gravidity, a to v důsledku toho, že v těhotenství se zvyšuje hladina vWF a aktivita ADAMTS13 se fyziologicky snižuje při jeho zvýšené konsumpci. Pokud je ale přítomen její kongenitální deficit, může se choroba snáze a rychleji manifestovat (8).
Vyšetření aktivity ADAMTS13 je klíčové pro stanovení diagnózy; aktivita pod 10 % potvrzuje diagnózu TTP. Znalost aktivity ADAMTS13 hraje zásadní roli v diferenciální diagnostice TMA, a proto je potřeba ji mít k dispozici co nejdříve. V současné době ji v ČR vyšetřuje již řada laboratoří ve statimovém režimu, a tak je možné mít výsledek dostupný do 24 hodin.
U pacientů s TTP bývá nejvíce postiženým orgánem mozek. Klinicky je pro manifestaci onemocnění typická horečka, neurologické příznaky (encefalopatie, křeče, známky CMP), významná trombocytopenie (často pod 30 x 109/l), MAHA a renální selhání. Ne všechny příznaky musí být plně vyjádřeny u všech případů, zejména renální postižení může být mírné či zcela chybět. V rámci rozlišení TTP od jiných příčin TMA nám může pomoci PLASMIC či modifikované francouzské (French) skóre (viz Tab. 1), která ale nebyla validována pro těhotné pacientky.
Léčba TTP patří jednoznačně do rukou hematologů. Mezi základní opatření patří snaha o normalizaci hladin ADAMTS13. Toho lze dosáhnout u kongenitálních forem TTP podáváním mražené plazmy či prováděním výměnných plazmaferéz (PLEX – plasma exchange). Jejich smysl je zde nejenom v tom, že můžeme podat větší objem mražené plazmy jako substituci chybějícího ADAMTS13, ale také odstranit z cirkulace multimery vWF, které jsou základní komponentou agregátů s trombocyty. Novou možnost pak představuje rekombinantní ADAMTS13, který se dostává do klinické praxe zvláště u opakovaně relabujících pacientů. U imunitně navozené TTP se léčba zaměřuje na blokádu tvorby protilátek proti ADAMTS13 imunosupresivy a jejich odstranění pomocí PLEX, kdy je plazmou jako náhradním roztokem navíc doplňována ADAMTS13. Základem imunosupresiv jsou u gravidních žen kortikosteroidy, azathioprin či kalcineurinové inhibitory (cyklosporin A, takrolimus). I když všechny tyto léky jsou během gravidity poměrně bezpečné, měli bychom při stanovení této diagnózy během těhotenství zvážit jeho co nejrychlejší ukončení (samozřejmě s ohledem na vyzrálost plodu). Rituximab, monoklonální protilátka proti CD20 receptoru na B lymfocytech, která se mimo graviditu podává po selhání kortikoidů nebo již v první linii v kombinaci s kortikoidy, má být v těhotenství s ohledem na nedostatek důkazů o bezpečnosti podána, pokud možný prospěch převáží riziko. Kojení se u žen léčených rituximabem nedoporučuje. Podobná pravidla platí pro podávání humanizované protilátky proti doméně A1 vWF caplacizumabu v graviditě a při kojení (3).
Diagnóza PE zahrnuje nový vznik hypertenze po 20. týdnu těhotenství (systolický TK ≥ 140 mmHg a/nebo diastolický TK ≥ 90 mmHg), rozvoj proteinurie a/nebo projevy orgánové dysfunkce a/nebo vývoj fetální růstové restrikce (fGR). Nemalé počty těhotných vyvinou PE z gestační hypertenze. Ta, na rozdíl od PE, není mezi TMA zařazována a ani u ní ve většině případů známky TMA neidentifikujeme. V obou případech se jedná o stavy, které lze považovat z větší části za reverzibilní. Původ PE je třeba hledat již v samém úvodu těhotenství – chybnou komunikací imunitního systému matky s trofoblastem. Až velmi pozdním důsledkem chybné „materno-fetální“ komunikace je abnormální remodelace spirálních arterií deciduy a rozvoj endotelové dysfunkce, charakterizované mimo jiné výkyvy hladin antiangiogenních (sFlt-1, s-eng) a angiogenních (PlGF, PAPP-A) látek. Tyto parametry jsme schopni identifikovat již od 10. týdne gravidity a změny jejich hladin/poměru se mohou používat k predikci PE v klinické praxi (Tab. 1) (9). Poměrně recentní studie PRAECIS (10) se snažila validovat tento index a ukázala, že poměr sFlt1:PlGF ≥ 40 signalizuje rozvoj relativně závažného průběhu PE v následujících 2 týdnech s vysokou senzitivitou. Samotné toto kritérium ale nestačí pro stanovení diagnózy PE a naopak, nižší hodnota její rozvoj zcela nevylučuje (11). Endotelová dysfunkce se týká jak systémového oběhu matky, tak i mikrocirkulace placenty.
Obecně platí, že dobře korigovaná gestační hypertenze s sebou nese možnost ambulantního sledování za pravidelné kontroly laboratorních ukazatelů funkce ledvin, jater, iontogramu, základních koagulačních testů, krevního obrazu, dále selfmonitoring krevního tlaku a pravidelné hodnocení růstu plodu. Při dobré kompenzaci TK a absenci růstové restrikce plodu preferujeme porod do termínu a plánování porodu podle daných podmínek po dosažení termínu. V případě PE již volíme hospitalizaci; cílem našeho managementu jsou identická opatření jako při gestační hypertenzi, a při absenci ohrožení matky a/nebo plodu se snažíme prodloužit těhotenství. V případě progrese PE do obrazu onemocnění „s těžkými rysy“ (TK ≥ 160/110 mmHg, známky TMA či orgánové dysfunkce apod.), plánujeme porod dle daných podmínek v nejkratší možné době. Samotná PE ale nepředstavuje kontraindikaci vaginálního vedení porodu.
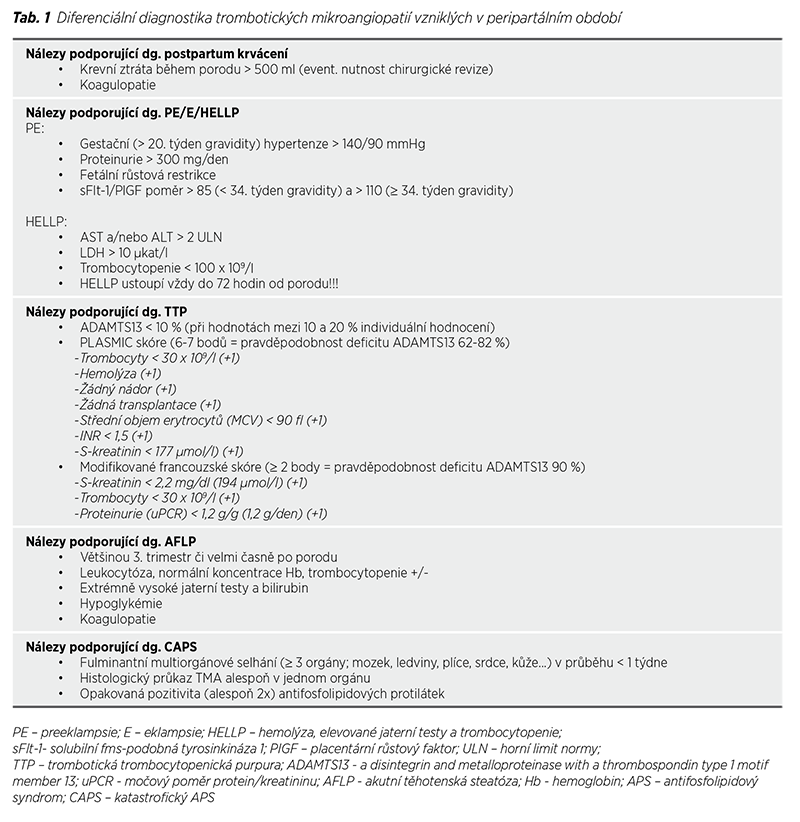
Podle dostupných poznatků je zjevné, že i HELLP syndrom může být spojen s mutacemi genů kódujících proteiny komplementu a řadí se mezi TMA (12). HELLP syndrom je pracovní diagnóza, jejíž definitivní potvrzení můžeme stanovit až odezní-li projevy a laboratorní příznaky TMA do 48-72 hodin po porodu. Diagnostická kritéria HELLP syndromu jsou laboratorní, a nejčastěji se závažnost onemocnění klasifikuje podle Mississippi klasifikace (Tab. 2). Hemolýza se u HELLP syndromu projevuje MAHA, tzn. zvýšením aktivity LDH provázeným poklesem koncentrace haptoglobinu (event. hemopexinu) a zvýšením koncentrace nekonjugovaného bilirubinu. Elevace transamináz se týká především AST (hepatocelulární poškození), ale obvyklý je i záchyt elevace ALT. Trombocytopenie je ale nejvýznamnějším prognostickým ukazatelem diagnózy. Dalšími doprovodnými laboratorními nálezy mohou být elevace CRP (v rámci systémové zánětlivé odpovědi) a patologie koagulačních testů včetně zvýšení koncentrace D-dimerů a event. pokles aktivity antitrombinu (v rámci rozvíjející se konsumpční koagulopatie) a zvýšení koncentrace vWf (obraz endotelové dysfunkce). Pokles eGFR (odhadovaná glomerulární filtrace) není pro diagnózu typický.
Také klinické projevy jsou velmi nespecifické a zahrnují kromě bolestí v epigastriu, nauzey a/nebo zvracení také „flu-like“ obtíže připomínající virózu, a to včetně subfebrilií. Klinické obtíže mají zpravidla progresivní charakter. Na druhou stranu, minimálně v počátcích HELLP syndromu nemusí být přítomny. Nemalý počet těhotných vyvine obraz HELLP syndromu z již diagnostikované gestační hypertenze/PE. HELLP syndrom komplikuje 0,5-1 % těhotenství (13). Je důležité vědět, že 1/3 žen vyvine diagnózu do 48 hodin po porodu. Právě skupina žen, u nichž se vyvine HELLP syndrom až po porodu, vyžaduje velmi bedlivé sledování, protože může zahrnovat i pacientky, u nichž se o HELLP syndrom nejedná a může jít o obraz jiných zde uvedených TMA. Zdá se, že v diferenciální diagnostice mezi HELLP syndromem a CM-HUS by nám mohla pomoci kombinace laboratorních testů LDH a sérového kreatininu (14). Ve studii srovnávající 46 pacientek s CM-HUS vzniklým v souvislosti s graviditou a 45 nemocných s HELLP syndromem se ukázalo, že je-li 72 hodin po porodu sérový kreatinin nad cca 170 μmol/l a LDH nad 10 μkat/l, pak je více jak 95% pravděpodobnost, že se jedná o CM-HUS.
Základním terapeutickým postupem v léčbě PE/HELLP syndromu je porod. Obecně platí, že by u PE/HELLP syndromu mělo dojít ke klinické a laboratorní regresi nálezů nejpozději do 72 hodin po porodu. Tento stav je ale většině porodníků v ČR dobře znám, a to i díky řadě českých publikací (15).
Pacientkám s proběhlou PE/HELLP po porodu doporučíme dispenzární péči praktickým lékařem a po skončení šestinedělí je vhodné verifikovat, zdali nepřetrvává proteinurie či dysfunkce ledvin. Tyto pacientky jsou ve zvýšeném riziku rozvoje stejné komplikace i během následujících gravidit.
CM-HUS je život ohrožující onemocnění, které je způsobeno TMA vyvolanou nadměrnou aktivací alternativní cesty komplementu. Sekundární formy HUS bývají naproti tomu spojeny s primárním poškozením endotelu, které způsobuje aktivaci komplementu až druhotně (viz výše). Všechny formy HUS mohou vést k ischemii řady orgánů s jejich následnou dysfunkcí a mohou také komplikovat probíhající graviditu.
CM-HUS zahrnuje dva základní typy onemocnění – geneticky vázané a získané. Geneticky vázané formy způsobují mutace v genech pro proteiny regulující aktivaci komplementu a vedou buď k funkčnímu deficitu těchto proteinů, nebo poruše jejich syntézy. Genetický původ onemocnění jsme schopni prokázat u zhruba 60 % případů (16,17). Mezi regulátory komplementu, které bývají nejčastěji postiženy, patří komplementární faktor H (CFH), komplementární faktor I (CFI), membránový kofaktorový protein (MCP) a trombomodulin (THBD). Z aktivátorů komplementu pak mutace nejčastěji postihují komplementární faktor B (CFB) a C3 složku komplementu (C3). Získané formy CM-HUS jsou charakterizovány vznikem protilátek proti některým komplementárním faktorům (nejčastěji proti CFH), což ve svém důsledku opět vede k jejich porušené funkci. Pro všechny formy CM-HUS je charakteristická primární porucha komplementu s jeho dysregulací.
Klinická manifestace CM-HUS zahrnuje renální dysfunkci (až renální selhání) ve většině případů, často s těžším průběhem než u TTP, postižení centrálního nervového systému, srdce či gastrointestinálního traktu. Jeho postižení se může projevovat jako pankreatitida, často mají nemocní i průjmy. To nezřídka vede v prvním kroku k diagnóze STEC-HUS, pro který je právě anamnéza těžkých, někdy i krvavých průjmů, „typická“. Během první ataky onemocnění CM-HUS je riziko rozvoje ESKD či smrti 33-40 %.
Riziko trvalého poškození ledvin, selhání ledvin či úmrtí během prvního roku od stanovení diagnózy se bez léčby pohybuje až kolem 65 % (18,19).
Těhotenství, a zejména pak porod, patří mezi spouštěče tohoto onemocnění, a tak lze očekávat relativní nárůst incidence CM-HUS u těhotných/rodiček. Řada těchto pacientek byla historicky označována jako pacientky s „atypickým HELLP syndromem“, protože k regresi laboratorních a klinických příznaků po 48-72 hodinách od porodu nedocházelo. V nemalém počtu případů tak s sebou takové situace nesly i úmrtí pacientek a přinejmenším i dlouhodobou morbiditu. Na rozdíl od HELLP syndromu (zde je především patrné postižení jater), bývají klinické příznaky orgánové dysfunkce u CM-HUS více vyjádřeny (viz níže). Nicméně, jedná se o soubor nespecifických projevů a konečná diagnóza CM-HUS se stejně musí učinit až po vyloučení jiných příčin TMA. Incidence CM-HUS se pohybuje kolem 1-2/milion obyvatel, u těhotných je to pak asi 1 případ na 25 000 gravidit, přičemž zhruba 2/3 se rozvíjí postpartum. Výskyt CM-HUS během gravidity je jednoznačně spojen se zvýšenou mateřskou i fetální mortalitou, rizikem předčasného porodu či hypotrofie plodu, a dále s rozvojem DIC (diseminovaná intravaskulární koagulopatie) či CMP (cévní mozková příhoda).
Dlouhou dobu se v léčbě CM-HUS, ale i sekundárních forem HUS, používalo podávání mražené plazmy či PLEX, jejichž cílem bylo zastavit aktivaci komplementu a zvýšit hladinu chybějících komplementárních faktorů s regulující funkcí. Účinnost této terapie je ale omezená a řada nemocných zůstávala závislých na dialyzačním léčení či měla jiné chronické orgánové poškození. Ekulizumab je první monoklonální protilátka proti C5 složce komplementu, která účinně blokuje terminální fázi aktivace alternativní cesty komplementu a zabraňuje vzniku C5a a C5b-9 komplexu. Proběhlé studie u nemocných s CM-HUS ukazují na velmi rychlý nástup účinku ekulizumabu s vymizením projevů TMA a zlepšením renálních funkcí (20,21). Nevýhodou léčby je nutnost podávat infúzi s ekulizumabem jedenkrát za 14 dní, což vede u řady nemocných ke snížené complianci k léčbě po vymizení akutních příznaků onemocnění. I když máme zatím omezená data ohledně podávání této léčby u těhotných pacientek, zdá se, že by mohla být bezpečná (22). V malém množství lék přechází přes placentu, zatímco v mateřském mléce detekován nebyl. Vzhledem k této velmi efektivní léčbě u do nedávné doby špatně ovlivnitelné choroby bychom se měli snažit ji co nejdříve odlišit od jiných forem TMA v graviditě, aby prodleva mezi diagnostikou a zahájením léčby byla co nejkratší.
Novější možností v léčbě CM-HUS je ravulizumab, který funguje stejným mechanizmem jako ekulizumab, ale úpravou molekuly došlo k tomu, že je možné ho podávat jen jedenkrát za 8 týdnů. U tohoto léku ale zatím máme velmi omezená data týkající se podávání v těhotenství a během kojení. Vzhledem k tomu, že oba léky blokují terminální fázi aktivace alternativní cesty komplementu, která hraje důležitou roli v ochraně organizmu před některými infekcemi, je nutné, aby nemocní před zahájením léčby absolvovali vakcinaci proti meningokokové meningitidě (minimálně 14 dní před aplikací první dávky léku). Pokud musí být léčba ekulizumabem či ravulizumabem zahájena ihned a nelze čekat 14 dní na vytvoření protilátek, vakcinace proběhne a spolu s ní se nemocní zajistí profylaktickou antibiotickou léčbou (cílenou na meningokoka), která trvá do rozvinutí efektu vakcinace.
AFLP je nově také řazena mezi TMA, i když projevy TMA zde bývají méně vyjádřené. Je to velmi závažná diagnóza charakterizovaná dysfunkcí nebo selháním jater těhotné/rodičky, která může vést k ohrožení života matky a plodu, včetně smrti. Maternální mortalita se v současné době pohybuje kolem 4 %. Incidence onemocnění se odhaduje přibližně na 1 na 7 000-20 000 těhotenství (23). AFLP se rozvíjí nejčastěji ve 3. trimestru (mezi 30.-38. týdnem gravidity).
Etiopatogeneze není přesně známa, předpokládá se abnormální metabolizmus mastných kyselin na straně plodu. Přibližně 20 % případů má doloženo deficit fetálního „long-chain 3-hydroxyacyl CoA dehydrogenase“ (LCHAD), jednoho z enzymů účastnícího se oxidace mastných kyselin (24,25).
Vzhledem k tomu, že u většiny pacientů není možné deficit enzymů beta oxidace mastných kyselin prokázat, zatím není zřejmé, jakými mechanizmy porucha ovlivňuje matku, ale obecně se předpokládá toxické ovlivnění hepatocytů matky intermediárními produkty z fetální cirkulace. Rizikovými faktory pro AFLP je mužské pohlaví plodu, vícečetné těhotenství, nízký body mass index < 20, již diagnostikovaná PE a předchozí AFLP. Klinicky se pacientky prezentují pod obrazem fulminantního jaterního selhání, se všemi souvislostmi s tím spojenými. Často je zjevný ikterus, únava, v anamnéze je polydipsie, polyurie. V laboratorních nálezech bývá přítomen „neúplný“ obraz HELLP syndromu, s trendem k poklesu počtu trombocytů. Jak je uvedeno níže, trombocytopenie není součástí diagnostických kritérií AFLP. Mnohdy se pacientky dostaví pro absenci vnímání pohybů plodu a může být potvrzeno intrauterinní fetální úmrtí. Podobně jako jiné TMA, především CM-HUS, je diagnóza AFLP stanovena často až „per exlusionem“, nicméně pro velmi vysoké riziko ohrožení života matky je naléhavě nutné pokusit se k diagnóze dospět co nejdříve. Pro určení pracovní, a posléze i definitivní, diagnózy je doporučeno využít tzv. Swansea kritéria (26). Splňuje-li pacientka 6 a více kritérií, potvrzuje to diagnózu AFLP (Tab. 3). Zobrazovací metody (USG, CT či MRI jater) nejsou pro stanovení diagnózy příliš přínosné.
Management AFLP je založen především na promptním ukončení těhotenství. Matka je při jaterním selhání nejvíce ohrožena koagulopatií typu DIC s neschopností jater adekvátně syntetizovat koagulační faktory. Jako u jiných TMA, je nutná mezioborová spolupráce s tím, že mezi život zachraňující opatření se u AFLP řadí substituce koagulačních faktorů, tj. především podávání plazmy a fibrinogenu. Důležitá je také korekce hypoglykémie a případných iontových změn.
Vaginální porod není kontraindikovaný, ale jeho umožnění závisí na stavu matky a plodu. Většinou se ale dává přednost císařskému řezu (SC). Důležité je mít na paměti, že se zde může rychle rozvinout těžká koagulopatie, proto je před provedením SC užitečné znát aktuální výsledky koagulačních testů (PT, APTT, fibrinogen), event. viskoelastických metod (rychleji dostupný komplexní pohled na hemostázu). Významnou poruchu koagulace je nutné korigovat (dominantně hypofibrinogenémií) a zajistit bezpečný počet trombocytů (při vaginálním porodu nebo porodu SC nad 50 x 109/l, při epidurální anestezii nad 80 x 109/l).

APS je autoimunitní syndrom charakterizovaný arteriálním a/nebo venózním tromboembolizmem a/nebo těhotenskou morbiditou s trvale pozitivními antifosfolipidovými protilátkami (APLA protilátky). Mezi APLA protilátky patří: lupus antikoagulant (LA), antikardiolipinové protilátky (ACLA) a protilátky proti beta2-glykoproteinu 1 (anti-B2GPI). Neexistují jednotná diagnostická kritéria, jen kritéria klasifikační, která nám mohou pomoci s klasifikací onemocnění. Nejčastěji se dosud používala Sapporo kritéria (Tab. 4) (27), aktuálně jsou živě diskutována recentní ACR/EULAR kritéria z roku 2023 (28). Mezi další příznaky APS patří mikrovaskulární projevy (livedo racemosa, livedoidní vaskulopatické léze, akutní nebo chronická nefropatie, difuzní alveolární hemoragie, resp. kardiomyopatie nebo adrenální hemoragie), změny srdečních chlopní (ztluštění nebo vegetace) a hematologické změny (trombocytopenie). Velká část pacientek má těžkou hypertenzi. APS se v 50 % vyskytuje jako primární onemocnění, v ostatních případech jde o sekundární APS, který doprovází jiné autoimunitní choroby (především SLE).
Mezi klasifikační kritéria těhotenské morbidity je dle ACR/ EULAR kritérií řazeno (28):
• jinak nevysvětlené 3 a více po sobě jdoucí časné aborty před 10. týdnem gravidity,
• jinak nevysvětlené úmrtí plodu/potrat mezi 10. až 34. týdnem gravidity bez těžké PE nebo placentární insuficience,
• těžká PE a/nebo placentární insuficience do 34. gestačního týdne s nebo bez úmrtí plodu,
• těžká arteriální hypertenze,
• poruchy CNS – nově vzniklá bolest hlavy nereagující na léky, která není vysvětlena alternativní diagnózou, poruchy vizu,
• plicní edém, hepatopatie (abnormálně zvýšené koncentrace jaterních enzymů v krvi nebo silná přetrvávající bolest v pravém horním kvadrantu nebo v epigastriu nereagující na léky, kterou nelze vysvětlit alternativní diagnózou), renální dysfunkce (koncentrace sérového kreatininu > 97 μmol/l nebo zdvojnásobení jeho koncentrace při absenci jiného onemocnění ledvin),
• trombocytopenie pod 10 x 109/l,
• placentární dysfunkce, oligohydramnion, růstová retardace plodu, abnormální nebo neuspokojivý výsledek testů sledování plodu, abnormální křivka dopplerovské průtokové velocimetrie, cévní malperfúze matky na základě histologického vyšetření placenty.

CAPS (katastrofický APS) představuje život ohrožující formu APS s rychlým nástupem příznaků s mnohočetnými trombózami na úrovni makro- i mikrocirkulace („thromboinflammatory storm“) vedoucí k multiorgánovému postižení/ selhání (Tab. 5). Komplikuje asi 1 % všech APS. CAPS může být i první manifestací APS. V 70 % bývají postižené ženy. Postižen může být kterýkoliv orgánový systém (74 % ledviny, 56 % CNS, 55 % plíce – ve formě syndromu akutní dechové tísně či difuzního intraalveolárního krvácení). Intenzita, dynamika rozvoje a rozsah postižení orgánů odlišuje CAPS od APS.
Patofyziologicky APLA protilátky zvyšují přítomnost trombóz několika způsoby. Jde především o inhibici antikoagulační kaskády a fibrinolytické aktivity, zvýšení aktivace trombocytů a zvýšení aktivity komplementu (29). Pozitivita všech tří typů APLA protilátek (LA, ACLA a anti-B2GPI) bývá spojeno s výrazně závažnějším průběhem gravidity a vyšším rizikem ztráty plodu, než je tomu u pacientek s pozitivitou jen jedné z nich (30).
Mezi nejčastější „spouštěcí“ rizikové faktory rozvoje APS patří infekce, chirurgický výkon, nádorové onemocnění, rozvoj SLE či neadekvátní/neúčinná antikoagulační léčba u známého APS. U žen to pak mohou být hormonální změny během těhotenství a v postpartálním období, ale třeba i zahájení terapie hormonální antikoncepcí.

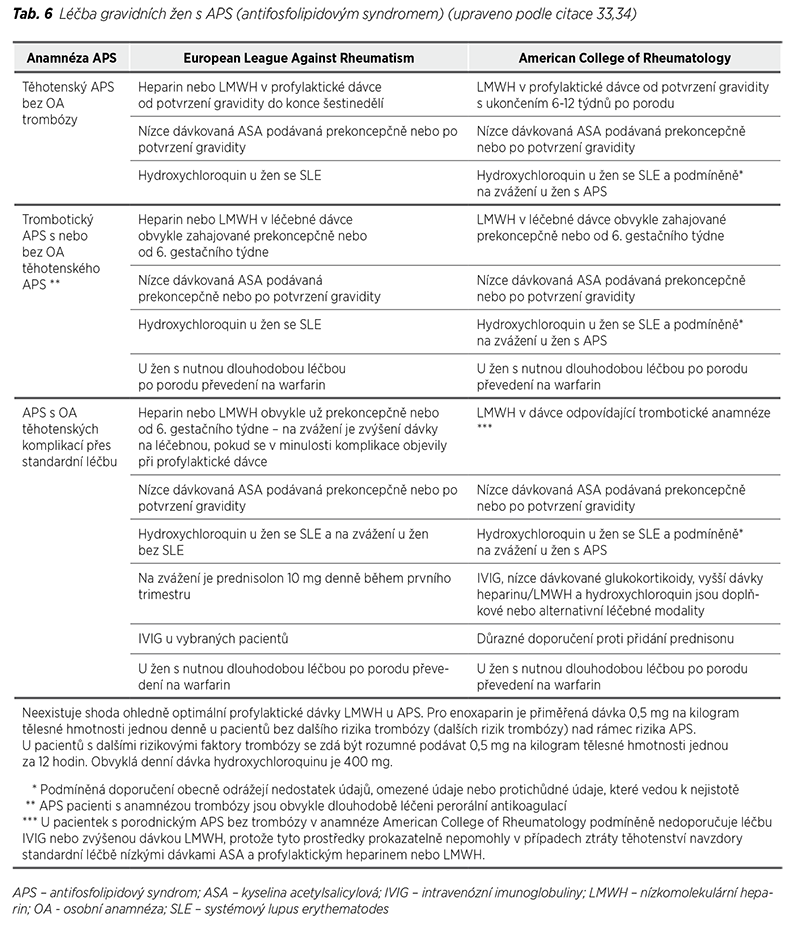
Rozhodování o antitrombotické profylaxi těhotenské patologie u APS nebo trombózy v graviditě nebo šestinedělí závisí na osobní anamnéze a laboratorních kritériích APS. Ženy s anamnézou trombózy, SLE, nevysvětlených těhotenských ztrát nebo předčasného porodu pro těžkou preeklampsii jsou ve vysokém riziku rekurence těchto příhod i přes standardní léčbu kombinací heparinu s nízce dávkovanou acetylsalicylovou kyselinou a nezávisle na typu nebo kombinaci přítomných APLA protilátek (31).
Riziko výskytu trombotických komplikací u matky se zdá být nižší při profylaxi kombinací heparinu s nízce dávkovanou acetylsalicylovou kyselinou, která ale neeliminuje riziko preeklampsie a placentární insuficience (32). Přehled doporučení léčby žen v graviditě a po porodu je uveden v Tabulce 6 (33,34).
Rozhodnutí o zahájení léčby CAPS je závislé na klinickém stavu pacientky a znalosti laboratorních výsledků. Typická „triple“ terapie CAPS zahrnuje antikoagulaci, glukokortikoidy a PLEX nebo vysokodávkované imunoglobuliny (400 mg/kg po dobu 5 dnů) (35,36). PLEX preferenčně používáme u těžké trombocytopenie a renální dysfunkce, resp. známek TMA; nejčastěji provádíme 5 procedur během 5 dnů a dále dle vývoje stavu. Velké krvácení, trombocytopenie a plná antikoagulace nejsou kontraindikací k provádění PLEX. U refrakterních pacientů lze zvážit léčbu rituximabem či ekulizumab (37). U nemocných s koincidencí APS a SLE je vhodné podávat hydroxychlorochin.
Klinické a laboratorní projevy TMA v graviditě a postpartum mohou zahrnovat širokou škálu symptomů a postižení různých orgánů a systémů, z nichž nejčastější jsou:
• CNS: zmatenost, mozkový infarkt, epileptické záchvaty, parestezie, parézy a plegie,
• renální: zvýšená hladina sérového kreatininu, hypertenze, snížená eGFR,
• krev: trombocytopenie, anémie, zvýšení aktivity LDH provázené snížením koncentrace haptoglobinu, event. hemopexinu a zvýšením počtu schistocytů,
• zrakové: amaurosis fugax, dvojité vidění, neostré vidění,
• kardiovaskulární: ICHS, hypertenze, difuzní dyskineza,
• gastrointestinální: průjem, kolitida, nauzea/zvracení, pankreatitida, bolesti břicha, gastroenteritida, hepatopatie,
• plicní: dyspnoe, difuzní alveolární hemoragie, edém,
• sub-/febrilie, „flu-like“ příznaky.
S ohledem na funkci ledvin je důležité si uvědomit jednu skutečnost. V průběhu těhotenství přirozeně stoupá eGFR (vzestup cirkulujícího volumu a plazmy), a tak je obecně velmi důležité sledovat dynamiku hladin sérového kreatininu. Za jeho horní fyziologickou mez je v graviditě považována hodnota přibližně 80 μmol/l. Hodnocení změn hladin kreatininu a eGFR v průběhu těhotenství představuje citlivější ukazatel poškození mikrocirkulace než „tradičně“ uváděné hodnoty kyseliny močové. Progresivní vzestup těchto hodnot nad uvedenou mez může signalizovat rozvoj akutního poškození ledvin spojeného s TMA.
U nemocných s projevy TMA a podezřením na HELLP syndrom je nezbytnou nutností monitorovat denně až do případné normalizace:
• krevní obraz s diferenciálním rozpočtem, retikulocyty a mikroskopické hodnocení počtu schistocytů v krevním nátěru,
• urea, kreatinin, Na, K, Cl, kyselina močová, AST, ALT, ALP, GMT, bilirubin celkový, LDH, haptoglobin, CRP, glykémie,
• PT, APTT, fibrinogen, antitrombin, D-dimery.
Vždy z odběrů provedených před PLEX nebo podáním plazmy
• ADAMTS13 aktivita a při poklesu stanovení protilátek proti ADAMTS13,
• přímý antiglobulinový test (PAT) – Coombsův test,
• imunologický panel: IgG, IgA a IgM, C3, C4, ACLA a anti-B2GPI, ANAb (antinukleární protilátky), ds-DNA (protilátky proti dvoušroubovici DNA), ENA (extrahovatelný nukleární antigen), ANCA (protilátky proti cytoplazmě neutrofilních leukocytů), anti-GBM (protilátky proti glomerulární bazální membráně),
• LA,
• protilátky proti komplementárnímu faktoru H,
• sFlt-1/PlGF poměr,
• užitečné je archivovat zamrazené vzorky pro budoucí použití.
Možné provést i po zahájení PLEX nebo po podání plazmy
• PCR průkaz shiga toxinu ve stolici,
• exprese MCP na makrofázích,
• proteinurie či uPCR (protein/kreatinin),
• moč chemicky + sediment,
• kultivace moči, hemokultury.
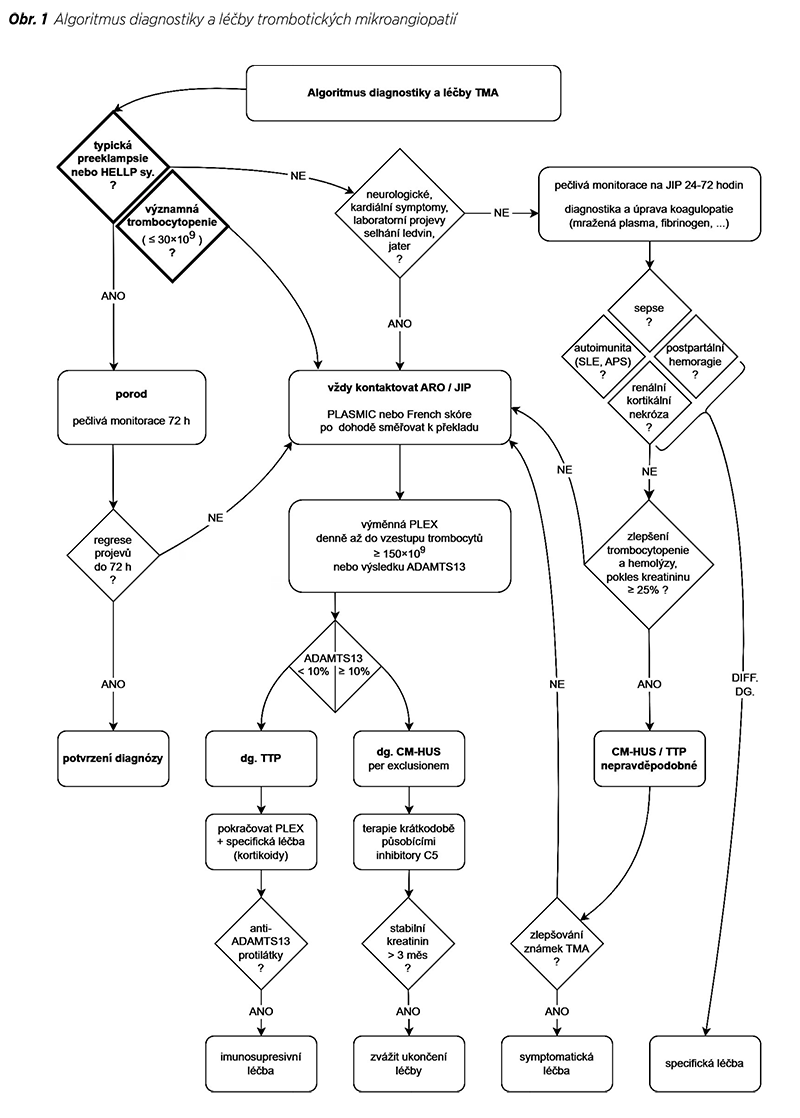
V rámci diferenciálně diagnostické rozvahy jednotlivých příčin TMA nám mohou pomoci některé skórovací systémy uvedené v tabulkách. Uvedený algoritmus vás pak provede jednotlivými diagnostickými kroky a usnadní vám orientaci v této nelehké problematice (Obr. 1).
Základním předpokladem úspěšného zvládnutí chorob asociovaných s TMA v graviditě a peripartálním období je především včas tuto jednotku odhalit a urychleně provést všechna vyšetření, která nám umožní v rámci diferenciální diagnózy co nejrychleji stanovit přesné onemocnění. V dnešní době máme kromě urychleného porodu k dispozici již i řadu nových terapeutických nástrojů, které ovlivňují samotné patofyziologické příčiny těchto stavů, a tak výrazně zvýší šanci na jejich úplnou, a hlavně rychlou úpravu. Významným způsobem tak snižují jak mateřkou, tak i novorozeneckou mortalitu.

